
Спінальна м’язова атрофія, СМА (Spinal Muscular Atrophy, SMA) — генетичне захворювання, що характеризується розвитком прогресуючої м’язової слабкості. У цій статті ми коротко ознайомимося із захворюванням СМА, причиною його виникнення, а також способами лікування.
Діагноз СМА – що це таке?
СМА хвороба – це орфанне захворювання, тобто таке, що зустрічається не частіше ніж 1 випадок на 2000 населення. Кожна 35 людина є безсимптомним носієм мутації, що призводить до СМА, і хвора дитина народжується, коли зустрічаються 2 таких мутації: з боку матері та з боку батька. Це відбувається приблизно 1 раз на 6000 народжень — у сім’ях, де ніхто, як правило, не чув про таку хворобу, де не було хворих родичів, шкідливих факторів середовища — нічого, що могло б навести на думку про високий ризик генетичних проблем.
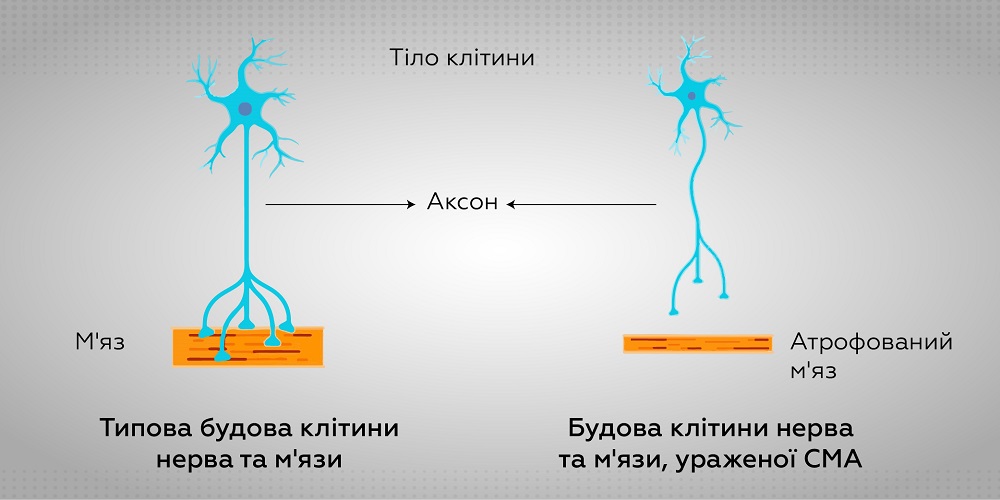
СМА виникає через мутацію гена SMN1, розташованого на 5 хромосомі, що призводить до порушення роботи рухових нейронів спинного мозку. В результаті розвивається м’язова слабкість кінцівок, що наростає, гіпотрофія м’язів. Крім того, захворювання також характеризується скелетними порушеннями (сколіозом, деформаціями грудної клітки), розвиваються контрактури, поступово розвивається дихальна недостатність. Інтелект при СМА залишається повністю збереженим та розвивається так само, як у здорових людей.
тіло клітини, аксон
Виділяють декілька основних типів СМА
СМА 1 типу
- СМА 1 типу (хвороба Вердніга-Гоффмана) починається у віці <6 місяців і характеризується найтяжчим протіканням захворювання: 95% таких дітей помирають протягом першого року життя. До 4 років вмирають всі діти, зазвичай від дихальної недостатності.
СМА 2 типу
- СМА 2 типу (хвороба Дубовиця) починається пізніше, у віці 7-18 місяців і характеризується більш повільним перебігом. До 2-3 років більшість малюків прикуті до інвалідного крісла. Діти часто вмирають у ранньому віці від дихальної недостатності. Ті, у яких прогресування м’язової слабкості зупиняється, виживають, але часто буває викривлення хребта.
СМА 3 типу
- СМА 3 типу (хвороба Кугельберга-Веландера) характеризується початком після 18 місяців. Діти вчаться самостійно сидіти та ходити, при цьому спостерігаються проблеми з більш складними руховими актами: бігом, підйомом сходами та іншими активностями. М’язова слабкість здебільшого прогресує повільно. Частина пацієнтів доживає до дорослого віку, зберігаючи здатність до пересування, інші до підліткового віку потребують інвалідного візка. Згодом можуть виникнути проблеми з ковтанням, відкашлюванням, диханням. При належному догляді такі пацієнти мають нормальну тривалість життя (тривалість життя залежить від часу розвитку порушень дихання).
СМА 4 типу
- СМА 4 типу, дуже рідкісний тип, коли захворювання проявляється вже у дорослому віці (зазвичай після 30 років). Наростання слабкості дуже повільне, м’язи, головним чином стегон і плечей, повільно слабшають та атрофуються. Тривалість життя не змінюється.

типи СМА
Як виникає SMA хвороба
Причиною виникнення та тяжкості прояву СМА є недостатня кількість SMN-білка (survival motor neuron: рухового (моторного) нейрона виживання) у клітинах. SMN білок забезпечує добрий стан рухових нейронів, відповідальних за передачу сигналів від мозку до м’язів. Якщо SMN білка мало або він відсутній, рухові нейрони вмирають і відмирають м’язи.
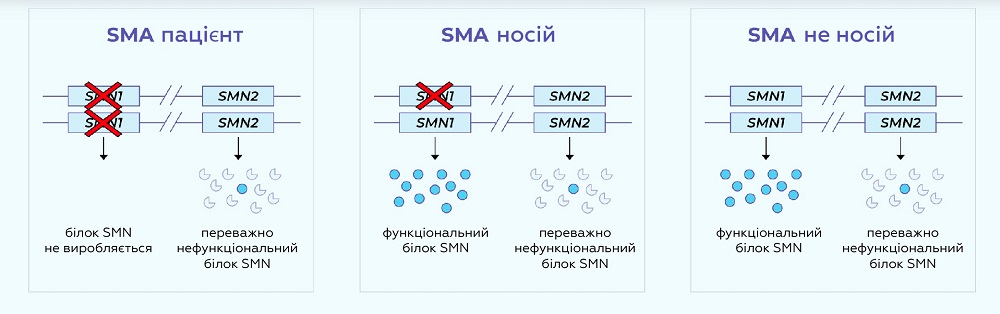
SMN білок виробляється переважно у SMN1 гені. У людей зі SMA немає функціональних копій гена SMN1 у якому білок SMN вироблявся б. Люди з однією копією SMN1 є безсимптомними носіями SMA (хоча пара носіїв може мати дітей з SMA).
СМА носій, пацієнт
Дуже схожий ген SMN2 теж істотно впливає на SMA. SMN2 генерує переважно нефункціональний білок SMN та невелику кількість функціонального SMN білка. Ця відмінність між стабільністю білків SMN1 та SMN2 є результатом одиночної нуклеотидної заміни (a C>T в екзоні 7).
Число копій SMN2 відрізняється у популяції. Для людей зі СМА чим більше копій SMN2 вони мають, тим менш сильно у них виявляються симптоми. Тому визначення кількості копій SMN2 є важливим для прогнозу хвороби.
На даний момент відомо, що тяжкість проявів залежить від кількості копій гена SMN2. Цей ген частково замінює вироблення білка SMN, якого бракує при СМА. Тому чим більше копій цього гена, тим пізніше захворювання починає проявлятися і тим повільніше розвиваються симптоми.
Генетичний аналіз СМА
Основним методом визначення СМА є генетичний аналіз, який спрямований на виявлення мутацій у гені SMN1. Діагноз СМА підтверджується, якщо виявлено делецію обох алелів SMN1.
Про лікування СМА та про те, яка ситуація з цим захворюванням в Україні, читайте у продовженні статті.