
Спинальная мышечная атрофия, СМА (Spinal Muscular Atrophy, SMA) — генетическое заболевание, характеризующееся развитием прогрессирующей мышечной слабости. В этой статье мы вкратце ознакомимся с заболеванием СМА, причиной его возникновения, а также способами лечения.
Диагноз СМА — что это такое?
СМА болезнь — это орфанное заболевание, то есть такое, которое встречается не чаще 1 случая на 2000 населения. Каждый 35-й человек является бессимптомным носителем мутации, приводящей к СМА, и больной ребенок рождается, когда встречаются 2 таких мутации: со стороны матери и со стороны отца. Это происходит примерно 1 раз на 6 000 рождений — в семьях, где никто, как правило, не слышал про такую болезнь, где не было больных родственников, вредных факторов среды — ничего, что могло бы навести на мысль о высоком риске генетических проблем.
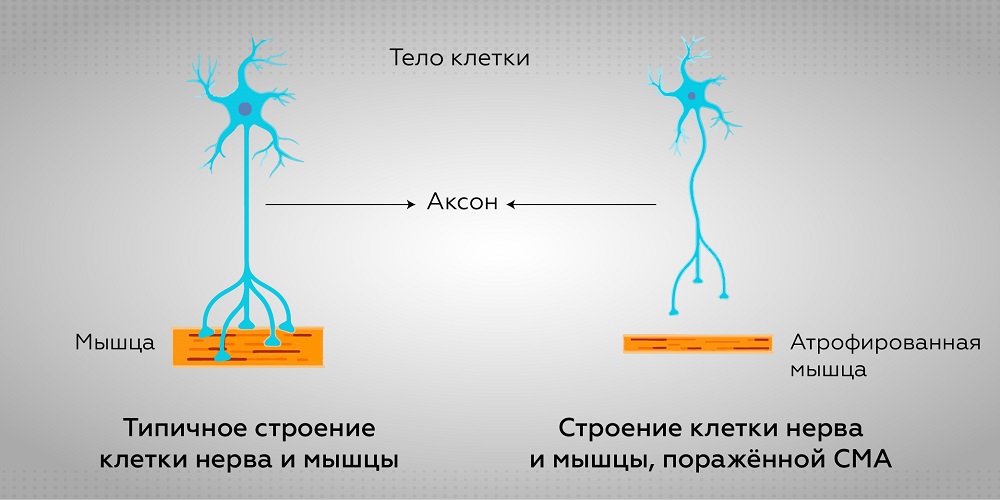
СМА возникает из-за мутации гена SMN1, расположенном на 5 хромосоме, которая приводит к нарушению работы двигательных нейронов спинного мозга. В результате развивается нарастающая мышечная слабость конечностей, гипотрофия мышц. Кроме того, заболевание также характеризуется скелетными нарушениями (сколиозом, деформациями грудной клетки), развиваются контрактуры, постепенно развивается дыхательная недостаточность. Интеллект при СМА остается полностью сохранным и развивается так же, как у здоровых людей.
тело клетки
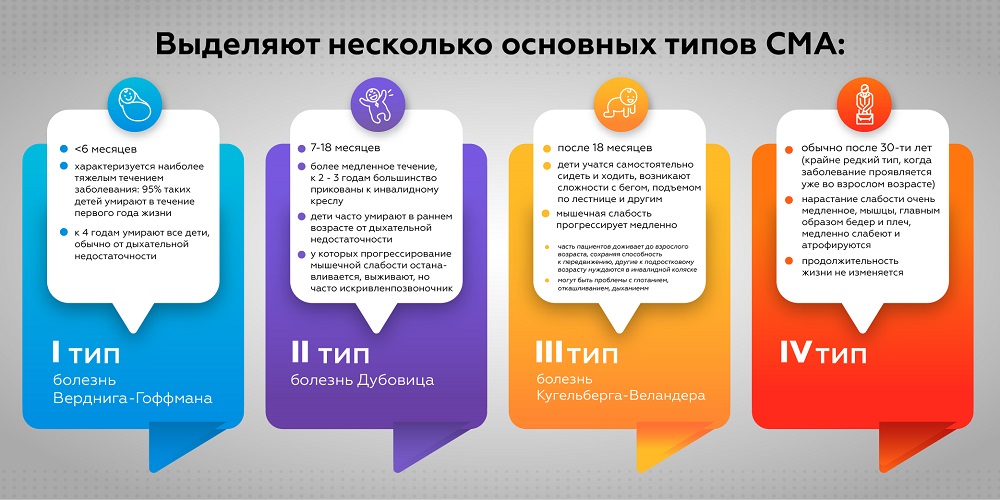
Выделяют несколько основных типов СМА.
СМА 1 типа
- СМА 1 типа (болезнь Верднига-Гоффмана) начинается в возрасте <6 месяцев и характеризуется наиболее тяжелым течением заболевания: 95% таких детей умирают в течение первого года жизни. К 4 годам умирают все дети, обычно от дыхательной недостаточности.
СМА 2 типа
- СМА 2 типа (болезнь Дубовица) начинается позже, в возрасте 7-18 месяцев и характеризуется более медленным течением. К 2 — 3 годам большинство малышей прикованы к инвалидному креслу. Дети часто умирают в раннем возрасте от дыхательной недостаточности. Те, у которых прогрессирование мышечной слабости останавливается, выживают, но у них часто бывает искривление позвоночника.
СМА 3 типа
- СМА 3 типа (болезнь Кугельберга-Веландера) характеризуется началом после 18 месяцев. Дети учатся самостоятельно сидеть и ходить, при этом наблюдаются проблемы с более сложными двигательными актами: бегом, подъемом по лестнице и другими активностями. Мышечная слабость в большинстве случаев прогрессирует медленно. Часть пациентов доживает до взрослого возраста, сохраняя способность к передвижению, другие к подростковому возрасту нуждаются в инвалидной коляске. Со временем могут возникнуть проблемы с глотанием, откашливанием, дыханием. При надлежащем уходе такие пациенты имеют обычную продолжительность жизни (продолжительность жизни зависит от времени развития нарушений дыхания).
СМА 4 типа
- СМА 4 типа, крайне редкий тип, когда заболевание проявляется уже во взрослом возрасте (обычно после 30-ти лет). Нарастание слабости очень медленное, мышцы, главным образом бедер и плеч, медленно слабеют и атрофируются. Продолжительность жизни не изменяется.
типы СМА
Как возникает SMA болезнь
Причиной возникновения и тяжести проявления СМА является недостаточное количество SMN-белка (survival motor neuron: двигательного (моторного) нейрона выживания) в клетках. SMN белок обеспечивает хорошее состояние двигательных нейронов, ответственных за передачу сигналов от мозга к мышечным. Если SMN белка мало или он отсутствует, двигательные нейроны умирают и мышцы отмирают.
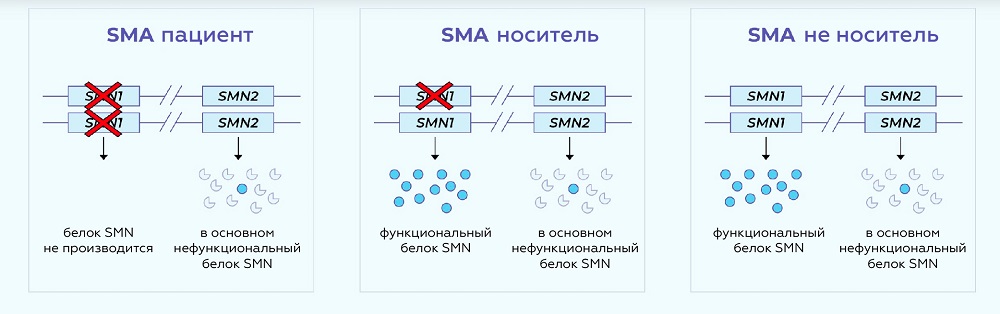
SMN белок вырабатывается преимущественно в SMN1 гене. У людей со SMA нет функциональных копий гена SMN1 в котором белок SMN вырабатывался бы. Люди с одной копией SMN1 являются бессимптомными SMA-носителями (хотя пара носителей может иметь детей со SMA).
СМА пациент, носитель
Очень схожий ген SMN2 тоже значительно влияет на SMA. SMN2 генерирует преимущественно нефункциональный белок SMN и небольшое количество функционального SMN белка. Это отличие между стабильностью белков SMN1 и SMN2 является результатом одиночной нуклеотидной замены (a C>T в экзоне 7).
Число копий SMN2 отличается в популяции. Для людей со СМА чем больше копий SMN2 они имеют, тем менее сильно у них проявляются симптомы. Поэтому определение количества копий SMN2 важно для прогноза болезни.
На настоящий момент известно, что тяжесть проявлений зависит от количества копий гена SMN2. Этот ген частично замещает выработку белка SMN, которого недостает при СМА. Поэтому чем больше копий этого гена, тем позже заболевание начинает проявляться и тем медленнее развиваются симптомы.
Генетический анализ СМА
Основным методом определения СМА является генетический анализ, который направлен на выявление мутаций в гене SMN1. Диагноз СМА подтверждается, если обнаружена делеция обоих аллелей SMN1.
О лечении СМА и о том, как обстоят дела с данным заболеванием в Украине, читайте в продолжении статьи.